1 概述
范科尼综合征(Fanconi syndrome)也称Fanconi-de Toni综合征、骨软化-肾性糖尿-氨基酸尿-高磷酸尿综合征、多种肾小管功能障碍性疾病。是指遗传性或获得性近端肾小管的功能异常引起的一组征候群。临床表现为肾性过多丢失而产生的全氨基酸尿、磷酸盐尿、葡萄糖尿、碳酸氢盐尿以及尿酸等有机酸尿;过多丢失电解质而产生的各种代谢性并发症,如低磷血症、低钙血症、高氯性代谢性酸中毒、维生素D缺乏病、骨质疏松、脱水、生长迟缓等;因丢失分子量小于5万的蛋白质而产生肾小管性蛋白尿。通过对氨马尿酸清除试验显示肾小管排泄功能障碍,肾小球功能基本正常或与酸中毒相比不成比例。近年有人将抗利尿激素抵抗的多尿症也包括在其中,但以氨基酸尿、糖尿、磷酸盐尿为基本诊断指标。
范科尼综合征国内罕见,婴儿与成人均可发病,起病缓慢,多于青壮年期出现症状,有肾性糖尿、多种氨基酸尿、高钙尿症、肾丢失钠、低磷血症、近端肾小管性酸中毒、低尿酸血症、肾小管性蛋白尿,低钾血症(肌无力、软瘫、周期性瘫痪等),低钙血症(手足搐搦症)等。常见并发症为肾小管性酸中毒;低钾血症;继发性甲状旁腺功能亢进、肾性骨病、骨畸形、骨软化症;肾结石等。本综合征病因复杂,预后与根底病息息相关,如及时采取适当治疗,一般预后尚好。但有些根底病无法根治,故预后不良,常因继发感染或肾功能衰竭而于儿童期死亡。
8 病因
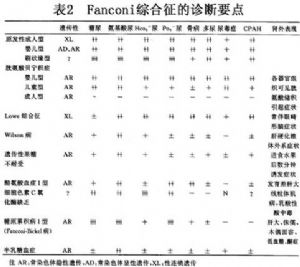
范科尼综合征的病因很多,可分为原发性与继发性两类。原发性Fanconi综合征又分为:婴儿型、成人型以及刷状缘缺失型三种类型。继发性Fanconi综合征又包括继发于遗传性疾病与继发于后天获得性疾病。前者包括:胱氨酸储积病、酪氨酸血症Ⅰ型、糖原贮积病ⅠⅠ型、半乳糖血症、遗传性果糖不耐受、细胞色素C氧化酶缺乏症、Wilson病、Lowe综合征、遗传性成骨不全、Alport综合征、先天性肾病综合征、维生素D依赖性佝偻依赖性佝偻病等;后者包括:肾病综合征、移植肾、急慢性间质性肾炎、多发性骨髓瘤肾病、舍格伦综合征、肾淀粉样变性、重金属中毒、药物(过期四环素、氨基糖类抗生素、6-巯基嘌呤、顺铂等)引起的肾损害、低钾性肾病、甲状旁腺功能亢进以及肿瘤相关性肾病等。病因详见表1。

9 发病机制
Fanconi综合征发病机制尚未完全阐明。目前认为不同于单项物质转运异常,即不是由于某种特异性的载体或受体缺陷所致。主要有两方面机制:
9.1 肾小管细胞膜有漏隙,不能使溶质充分再吸收
反漏的证据是肾性糖尿属A型,表明葡萄糖转运再吸收部位较少,磷酸盐、碳酸氢盐在滤过负荷减少的情况下仍有丢失。这表明它们的排泌是通过肾小管的泄漏。
9.2 肾小管内能量代谢不足,产生的能量难以支持正常转运
有些毒物或药物中毒以及遗传代谢疾病使某些代谢产物在肾小管内储积过多影响了细胞内氧化磷酸化过程,ATP生成不足,没有足够的能量支持肾小管转运物质。无论什么机制可最终导致多种物质转运异常。范可尼综合征是近曲小管多项转运缺陷病,包括氨基酸、葡萄糖、钠、钾、钙、磷、碳酸氢钠、尿酸和蛋白质。原发性者近端小管呈天鹅颈样变形。
10 范科尼综合征的临床表现
范科尼综合征较罕见,多于成年出现症状,有肾性糖尿、多种氨基酸尿、高钙尿症、肾丢失钠、低磷血症、近端肾小管性酸中毒、低尿酸血症、肾小管性蛋白尿,低钾血症(肌无力、软瘫、周期性瘫痪等),低钙血症(手足搐搦症)等。长期低钙血症,可引起继发性甲状旁腺功能亢进、肾性骨病。本病最突出的临床表现为小儿维生素D缺乏缺乏病和成人的骨软化症。继发性范可尼综合征的临床表现,基本上与原发性者同,但可有其根底疾病的临床表现。本综合征临床表现复杂,根据其临床类型分述如下:
10.1 原发性Fanconi综合征
原发性Fanconi综合征包括3种类型:
10.1.1 (1)成人型Fanconi综合征
10~20岁以后起病,有多种肾小管功能障碍,如全氨基酸尿、糖尿、磷酸盐尿、高血氯性酸中毒、低钾血症等。突出的症状是软骨病,少数病例可有酮症晚期可出现肾功能衰竭。
10.1.2 (2)婴儿型Fanconi综合征
多于6~12个月发病,多尿、烦渴、脱水、便秘、无力、拒食、发热,生长发育迟缓肾性氨基酸尿,可有抗维生素D佝偻佝偻病及严重营养不良现象。实验室检查呈低血钾、低血磷、低血钙及碱性磷酸酶增高、高氯性代谢性酸中毒、尿中可滴定酸及NH4可减少,尿糖微量或4~5g/d,血糖正常,急性起病者预后差,常死于尿毒症。慢性起病者多于2岁以后发病,症状较轻,突出表现为侏儒和(或)抗维生素D佝偻佝偻病。
10.1.3 (3)特发性刷状缘缺失型Fanconi综合征
1984年Manz等首次报道1例小儿由于近曲小管刷状缘完全缺失而引起Fanconi综合征,因为葡萄糖及各种氨基酸载运系统完全丧失,故这些物质的清除率近于肾小球滤过率。
10.2 继发性Fanconi综合征
继发性Fanconi综合征多有原发病,不同病因引起者表现各有不同。
10.2.1 (1)胱氨酸储积症
胱氨酸储积症又称Lignac-Fanconi综合征,系胱氨酸沉着于细胞溶酶体而表现为Fanconi综合征。正常人细胞内溶酶体是细胞内蛋白降解的部位,细胞内蛋白降解产生氨基酸通过溶酶体膜转输系统输入胞质而被再利用。本病因溶酶体内胱氨酸运载体有缺陷,使胱氨酸在溶酶体中储积,从而破坏了溶酶体的完整性,并可使具有破坏性的溶酶体酶漏至细胞浆,影响了功能。本病与胱氨酸尿症不同,后者是肾小管上皮转运胱氨酸障碍,只引起胱氨酸尿,前者则引起许多器官细胞内胱氨酸储积,肾脏是主要受累器官之一。
胱氨酸储积病所引起的Fanconi综合征不同于其他原因所致Fanconi综合征,常以失钾、脱水、多饮、渗透性利尿为突出表现。临床上可分3型:
①婴儿型或肾病型:胱氨酸沉积于各种组织溶酶体内,在白细胞内可能比正常大80倍,肾脏髓质可能沉着近100倍,因肾小管损伤出现各种症状,患儿多于6个月左右开始发病,多尿、烦渴、便秘、多饮、呕吐、拒食、消瘦、发育障碍,由于脱水有反复发热,可发生维生素D缺乏缺乏病及侏儒症。由于角膜、结膜的胱氨酸沉着而畏光,眼底周围色素脱失,可引起末梢视网膜病变。此外尚可表现为甲状腺功能低下、糖尿病、脾大、脑水肿、肌病等。肾小管功能障碍表现为肾浓缩功能障碍、氢离子排泄功能障碍,而至尿液不能酸化至pH 5.5以下,呈肾小管性酸中毒。
②儿童型或中间型:10岁左右发病,进展较慢,骨病不严重,无侏儒症。组织胱氨酸含量远较婴儿型为低,白细胞内胱氨酸含量为正常的30倍,也可表现为肾脏病变,甚至发展为尿毒症,骨骼畸形、畏光、视网膜病变、胱氨酸引起的脾大也可产生。Fanconi综合征表现不明显。
③成人型:无肾病表现,以其他器官功能障碍为主。成人型又可分为急性与慢性,前者与婴儿型类似,后者与儿童型类似。
通过骨髓片、白细胞、直肠黏膜中的结晶分析或裂隙灯检查角膜有胱氨酸结晶而诊断。婴儿型因患儿拒食引起饥饿性酮症加上肾小管性糖尿,易误诊为幼儿糖尿病,需提高警惕。
10.2.2 (2)Lowe综合征
Lowe综合征系Lowe 1952年首先报道,亦称眼-脑-肾综合征,临床表现特点为:
①眼症状:先天性白内障(双侧)伴有先天性青光眼(牛眼)、视力严重障碍、眼球震颤及畏光。
②脑症状:严重智力发育迟缓,肌张力低、腱反射减弱或消失,患儿常哭泣样尖叫。
③肾小管功能障碍:多组氨基酸尿、磷酸盐尿、碳酸氢盐尿、尿酸化功能差,尿中排出赖氨酸、酪氨酸为多。还可有肾小管性蛋白尿、后期可发生慢性肾功能不全。按自然发展可分为3期:婴儿期,以眼脑症状为主。表现为头颅畸形(长头,前额高出,鞍鼻,高腭弓等);儿童期,出现不完全Fanconi综合征,有肾小管性蛋白尿,严重磷酸盐尿可引起抗维生素D佝偻佝偻病或骨质疏松。一般情况下有较轻或无糖尿、失钾及多尿。常有脐疝、隐睾畸形,以及特殊的手指小关节炎;成人期,肾小管病症状消退,出现肾功能不全或营养不良,常合并肺炎而死亡。本综合征主要是对症治疗,如纠正肾小管性酸中小管性酸中毒,抗维生素D佝偻佝偻病的治疗等。无根治办法,预后不良。常因继发感染或肾功能衰竭而于儿童期死亡。
肝豆状核变性(Wilson病)系少见的隐性遗传性代谢性疾病。因为血浆铜蓝蛋铜蓝蛋白(ceruloplasmin)含量降低,铜氧化酶活性降低导致肠道大量吸收铜,大量铜沉积于肝、脑、角膜、肾小管引起相应的症状。铜沉积于脑及肝引起锥体外系神经症状及肝硬化,铜沉积于角膜引起Kayser-Fleischer环。铜沉积于近端肾小管及远端肾小管引起Fanconi综合征,可伴有重碳酸盐丢失和肾钙质沉着伴高钙尿症,肾小管性酸中毒,肾钙化,肾结石。
范科尼综合征可用青霉胺治疗,促进铜从尿中排出但停用后会复发。其他治疗如二巯丙醇(BAL)可增加铜排出,口服硫化钠可改善神经系统和肝症状,但肾小管病变无改善,骨化醇可治疗骨病变。
10.2.3 (4)遗传性果糖不耐受
遗传性果糖不耐受为常染色体隐性遗传的酶缺乏病。因肝、肾组织中缺乏1-磷酸果糖醛缩酶或者1,6-二磷酸果糖醛缩酶的活性下降,从而使1-磷酸果糖不能裂解而储积于细胞内产生病变,同时因不能产生ATP而影响细胞的能量代谢。若给患者输注果糖可产生Fanconi综合征复合肾小管功能障碍,若杜绝果糖则肾小管功能正常。本病的发病机制可能是由于肾皮质细胞内降解1-磷酸果糖的醛缩酶缺乏,小管上皮细胞内磷酸盐减少,对腺苷脱氨酶(ADA)的抑制作用减弱,以至ADA活性增强,使腺苷脱氨生成次黄苷(inosine),经核苷磷酸化酶作用生成次黄嘌呤,再由黄嘌呤氧化酶作用生成尿酸。故产生低磷高尿酸血症。另外,由于磷可增加肾皮质中ATP产生率,使果糖转化为α-磷酸甘油,当低磷血症时,ATP生成减少,也会影响到能量供应。由此可见1-磷酸果糖不是毒性代谢产物,它不抑制酶系统,而是由于磷的耗空使ATP及其他高能磷酸化合物在肾小管细胞某些位点的产生受到很大限制。
婴儿期因摄食乳糖无症状,当食用果糖或水果时急性发病,摄食后20~40min出现呕吐、腹泻、低血糖与高尿酸血症,2h后出现急性Fanconi综合征,乳酸性酸中毒、高胆红素血症、肝大。及时停止摄入果糖,治疗低血糖,病情可能缓慢逆转,否则可直接威胁生命。
10.2.4 (5)酪氨酸血症(tyrosinemia)
酪氨酸血症(tyrosinemia)是由于患者缺少对羟苯丙酮酸氧化酶(hydroxylphenyl puruvic acid oxidase)而导致酪氨酸代谢异常可引起Fanconi综合征。其特征是血中酪氨酸、苯丙氨酸、蛋氨酸、丙氨酸显著增加,其他氨基酸很少增加。在尿中排出酪氨酸、苯丙氨酸、蛋氨酸和对羟苯丙酮酸,对羟苯乙酸的酚酸代谢产物也增加。临床上本病分成两型:Ⅰ型酪氨酸血症即为暂时性高酪氨酸血症。若投以酪氨酸会发生肝肾功能复合损害,长期持续则肾皮质肾小管发生变性,肝硬化伴门脉高压和腹水。有的病例出现维生素D缺乏缺乏病,白内障形成或由于胰岛细胞肥大而引起低血糖症。Ⅱ型的特征为持续性高酪氨酸血症,病情持续发展,有严重智力障碍,皮肤异常、白内障、生长缓慢,而无明显的肝肾损害,其对羟苯丙酮酸氧化酶活性正常。
饮食治疗(如低酪氨酸、低苯丙氨酸饮食)可改善Ⅱ型患者病情,对Ⅰ型患者可减轻肾小管损害,但对严重肝损害无效。
10.2.5 (6)细胞色素C氧化酶缺乏症
细胞色素C氧化酶缺乏症可引起Fanconi综合征,这是因为肾小管上皮细胞线粒体中缺乏该酶而使电子传递链中ATP合成及氧化磷酸化过程障碍。患者多在出生后11~13周发病,主要表现为线粒体肌病,乳酸性酸中毒及肾性糖尿、氨基酸尿、磷酸盐尿等肾小管功能障碍。
10.2.6 (7)多发性骨髓瘤所致Fanconi综合征
多发性骨髓瘤可伴有肾淀粉样变性或轻链蛋白(κ或λ)引起肾小管损伤而致非遗传性继发性Fanconi综合征。临床特征为骨痛、肌无力、疲乏、贫血、骨软化症、假性骨折等,并有葡萄糖尿氨基酸尿、磷酸盐尿、肾性尿崩症、肾小管性酸中毒等肾小管功能不全的表现。其中Fanconi综合征为多发性骨髓瘤的伴发症状。
10.2.7 (8)毒性物质引起的Fanconi综合征
毒性物质可引起继发性Fanconi综合征。例如过期的四环素其降解产物具有肾小管毒性。其临床特征为肌病、眩晕、酸中毒、多尿、低钾血症。虽然停药后可能恢复,但有的病程可持续2年以上。
15 鉴别诊断
1.婴儿型Fanconi综合征鉴别诊断应注意其他原因所致的肾小管性酸中毒,肌无力症状或步态不稳类似神经系统病变或原发性肌病。也极似Fanconi综合征的婴儿型应注意区分。
2.成人因其他代谢性骨病引起骨质疏松伴肌病也类似Fanconi综合征;尿毒症患者可有葡萄糖尿或氨基酸尿而无低磷酸盐血症;Wilson病也会与运动系疾病相混淆。总之,复合性肾小管排出溶质过多必须寻找其原发疾病。
16 范科尼综合征的治疗
16.1 病因治疗
继发性Fanconi综合征应治疗基础疾病。Wilson病或重金属中毒可通过促进毒物排泄,遗传代谢病通过饮食管理减少代谢毒性物质沉积,减轻对肾小管的损害。胱氨酸储积症继发性Fanconi综合征,应给予低胱氨酸饮食及对症治疗。骨病变可用维生素D225万~50万U或维生素D332000~1000U或羟胆骨化醇200~5000U。有脱水及酸中毒应作相应处理。早期可用枸橼酸钾钠溶液10~15ml口服,3~5次/d。青霉胺可试用于消除胱氨酸,但不能减少细胞内胱氨酸沉着;Dithiothreitol(DDT)疗效欠佳,半胱氨酸效果较好。
16.2 对症治疗
(1)纠正酸中毒:根据碳酸氢根丢失情况补充碱剂,2~10mmol/(kg·d),可用重碳酸氢盐、枸橼酸盐、乳酸盐等,4~5次/d,分次给服,以血中碳酸氢盐水平恢复正常为标准。补钠可使低血钾加重,应注意检测;对已有低血钾者宜同时补钾2~4mmol/(kg·d)。若碱剂用量过大患者不能耐受时,可加用氢氯噻嗪(双氢克尿塞)2~3mg/(kg·d),它可使细胞外液缩减而促进碳酸氢根的再吸收,但应谨慎防止肾小球滤过率下降。
(2)纠正低血容量:Fanconi综合征常因多尿而致脱水,除了针对病因治疗外,应补足含盐的液体(包括钠、钾、钙等),可采用定时口服,必要时临时追加。
(3)纠正低磷血症:应用中性磷酸盐1~3g/d,分5次服用。如有腹泻或腹部不适可减量。注意补磷可加重低血钙与骨病,故应合用维生素D 5000U/d或1,25(OH)2D30.25~0.5μg/d,应从少量开始,逐渐加至足量。为防止肾钙化应监测尿钙排量,以不超过0.6g/d为标准。